![]()
|
|
|
LAB 5 BASIC CELL CULTURE TECHNIQUES I. INTRODUCTION Cells in culture or in vitro are a useful model for studying the activity of cells in the whole organism or in vivo. Ten years ago or so cell culture techniques were considered somewhat esoteric. Today because of our better understanding of cell nutrition, metabolism and general growth environment it has become a fairly routine procedure. Today's lab will consist of subculturing and quantifying a vertebrate cell line derived from one-day old zebrafish embryos and is called the ZF4 line. The ZF4 line is a fairly prototypical cell line and behaves very much like many of the mammalian lines that are available. The American Type Culture Collection in Rockville, Maryland is one of the major repositories for the large number of cell lines that are available. To obtain a given cell line you just send them the name of the cells you wish to culture along with $425.00 and it arrives a few days later frozen on dry ice. These cultures are then rapidly thawed and placed in their appropriate growth medium under sterile conditions, and if all works out well you have a cell line to work with. I received the ZF4 line in this manner last month. Some characteristics of the cell line are given below.
1. Name: ZF4
2. Growth Media: ATCC complete growth medium: The base medium for this cell line is ATCC- formulated DMEM:F12 Medium Catalog No. 30-2006. To make the complete growth medium, add 10% fetal bovine (not heat inactivated). NOTE: DO NOT USE Ca++/Mg++ or EDTA TO DISSOCIATE THE CELLS.
3. Atmosphere: 95% atmosphere, 5% carbon dioxide (CO2)
4. pH of Media: 7.25
5. Growth Temperature: 28 C
6. Antibiotics: Pen/Strep (5000U/5mg) 100U/ml Penicillin, 100ug/ml Streptomycin, note, do not use antifungals with this line
7. Rinse Buffer: Sterile PBS, or growth media without sera
8. Subculture Enzyme: 0.25X Trypsin (Gibco 1505-065), note no EDTA or Ca++/Mg++
9. Subcultivation Ratio: A subcultivation ratio of 1:2 to 1:4 is recommended with media renewal 2 to 3 times per week
I am assuming you know how to use the compound microscope. If you have any questions or need a review of its operation, feel free to ask. A specialized type of microscope that is used in cell culture work is called an inverted phase contrast microscope. These are expensive but easy to operate. The light source is located above the stage and the objective lenses are located below the stage.
The light source and phase contrast optics take advantage of the refractive indexes of live cells to give contrast to unstained specimens. We will be using this microscope to monitor our cultures. There are a number of different techniques available to perform routine cell culture work. Below are some procedures I have found to work well.
II. PROCEDURES
STERILE TECHNIQUES
Aseptic or sterile technique is the execution of tissue culture procedures without introducing contaminating microorganisms from the environment. In doing tissue culture work, 70% of the problems are due to a lack of good sterile technique. Microorganisms causing the contamination problems exist everywhere, on the surface of all objects and in the air.
A conscious effort must be made to keep them out of a sterile environment. Because many and sometimes awkward manipulations are required for various techniques, tissue culture media used are often supplemented with antibiotics. Antibiotics do not eliminate problems of gross contamination which result from poor sterile technique or antibiotic-resistant mutants. Autoclaving renders pipettes, glassware, and solutions sterile.
Nutrient medium cannot be autoclaved. The compounds in nutrient medium are destroyed by the heat of autoclaving. Medium must therefore be sterilized by passing it through a sterile filter small enough in pore size to hold back bacteria and mycoplasmas (Millipore Sterivex - GS 0.22u disposable filter units). Here are some rules of thumb to follow to keep your medium, cultures, and glassware from becoming contaminated:
1. Wipe your work area and hands with 70% ethanol before starting.
2. Never uncover a sterile flask, bottle, petri dish, etc., until the instant you are ready to use it. Return the cover as soon as you are finished. Never leave it open to the environment.
3. Sterile pipettes should never be taken from the wrapper until they are to be used. Keep your pipettes at your work area. Sterile pipettes do not have to be flamed. Pipetting your cells with a hot pipette will kill them.
4. When removing the cap from a bottle, flask, etc., do not place the cap with the open end upright on the lab bench. Do not hold the opening straight up into the air. If possible, tilt the container so that any falling microorganisms fall onto the lip.
5. Be careful not to talk, sing, or whistle when you are performing these sterile procedures.
6. Do not draw from a different bottles with the same pipette. Because such a pipette has been exposed; the chance for contamination is too great; use sterile pipette for each bottle -- especially when pipetting medium.
7. Techniques should be performed as rapidly as possible to minimize contamination.
You may find yourself involved in a procedure which these sterile technique "rules-of-thumb" do not cover. Therefore, you must constantly be aware that microorganisms are everywhere and take proper steps to keep them out of your cultures. When first developing your aseptic technique you must always be thinking of sterility. Eventually it will become second nature to you.
Mastering good aseptic technique will save you considerable frustration in the labs to follow. Furthermore, the same principles for good aseptic technique also minimize biohazard risk to the investigator when infectious organisms or dangerous chemicals are used.
"EYEBALLING" THE CULTURES
Before doing anything with a culture, its general "health" and appearance should be evaluated. This can be done quickly and quantitatively by making the following observations:
1. Check the pH of the culture medium by looking at the color of the indicator, phenol red. As a culture becomes more acid the indicator shifts from red to yellow-red to yellow. As the culture becomes more alkaline the color shifts from red to fuchsia (red with a purple tinge). As a generalization, cells can tolerate slight acidity better than they can tolerate shifts in pH above pH 7.6.
2. Cell attachment. Are most of the cells well attached and spread out? Are the floating cells dividing cells or dying cells which may have an irregular appearance?
3. Percent confluency. The growth of a culture can be estimated by following it toward the development of a full cell sheet (confluent culture). By comparing the amount of space covered by cells with the unoccupied spaces you can estimate percent confluency.
4. Cell shape is an important guide. Round cells in an uncrowded culture is not a good sign unless these happen to be dividing cells. Look for doublets or dividing cells. Get to know the effect of crowding on cell shape.
5. Look for giant cells. The number of giant cells will increase as a culture ages or declines in "well-being." The frequency of giant cells should be relatively low and constant under uniform culture conditions.
6. One of the most valuable guides in assessing the success of a "culture split" is the rate at which the cells in the newly established cultures attach and spread out. Attachment within an hour or two suggests that the cells have not been traumatized and that the in vitro environment is not grossly abnormal. Longer attachment times are suggestive of problems. Nevertheless, good cultures may result even if attachment does not occur for four hours.
7. Keep in mind that some cells will show oriented growth patterns under some circumstances while many transformed cells, because of a lack of contact inhibition may "pile up" especially when the culture becomes crowded. Get to recognize the range of cells shapes and growth patterns exhibited by each cell line.
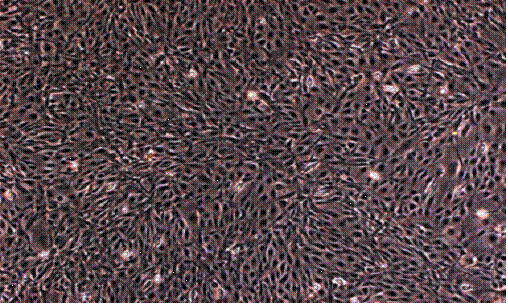
A confluent culture of ZF4 cells
SUBCULTURING PROTOCOL FOR ANCHORAGE-DEPENDENT CELLS:
1. Decant supernatant fluid from culture into a waste collection jar, applying the principles of sterile technique.
2. Optional: Rinse by adding 7 ml of PBS or growth media without sera to the T-25, then decant into waste collection jar.
3. Rinse by adding 3 ml of cold trypsin to the T-25, then decant into waste collection jar. This will remove the proteins present from the old media that tend to decrease the ability of the trypsin to act on the cells.
4. Now add 3 ml of cold trypsin and examine under low magnification for 3-7 minutes with the phase contrast inverted scope. When it appears that most of the cells have rounded up, but have not yet completely detached, whack the T-25 gently. Then immediately return to the hood and pipette the cells up and down several times to help break up the clumps of cells.
5. Remove 0.1 ml (100 ul) of cells in trypsin and place in microfuge tube for cell counting.
6. Add 9.0 ml fresh media to three new flasks (if you are doing a 1:3 split). Using the same pipette, and as continuation of the same process, draw up the cell suspension and quickly dispense a 0.9 ml aliquot into each T-25 flask containing 9.1 ml of media in the new flask giving a total volume of 10 ml.
7. Incubate at 28C and monitor periodically, beginning 30-45 minutes after inoculation. Rapid attachment (within 1 hour or so) is indicative, but not proof, that all went well.
8. Start making observations, such as those described in the handout "Eyeballing the Culture."
Directions for Use of Coulter Counter Particle Counter
1. Turn power switch to “I” position to turn instrument on
2. Lower specimen tray and replace blue “clenz” fluid with salt solution then raise up tray and place the aperture in salt solution
3. Press “functions” till F1 screen comes up then use arrow keys (>) to select “Prime Aperture”, then hit start, this takes you to F2 screen, then press start again
4. When finished, remove salt solution and replace with your sample in salt solution, be sure that the aperture is in sample
5. Press the “Setup” button – the “S1” screen should come up, then press “Start” to read your sample
6. Record your count and be sure to take into account the dilution factor, the instrument is set up to sample 0.5 ml and typically there is a total volume of 10 mls of sample.
7. To read another sample, just press start again
8. After every 10 counts flush the aperture again, this involves pressing “functions” till the F1 screen displays, then select “Flush Aperture”, then press start
9. At the end of the day do several counts (4-8) with blue Coulter “Clenz” solution, this is very important as it fills the aperture, pump and tubes in system with blue cleansingsolution and prevents lockups.
10. Every week replace blue Clenz fluid in cup and select “functions” then select “fill system”, this replaces fluid in system and prevents lockups
HEMOCYTOMETER COUNTING AND CELL VIABILITY (OPTIONAL)
The importance of accurate enumeration of cells at time of inoculation, at the termination of an experiment, and throughout the course of many experiments is self-evident. Cell enumeration with a hemocytometer is the most widely used method, and it continues to have a place in all laboratories, including those equipped with electronic cell counters (like us). This laboratory exercise will serve to introduce the use of a hemocytometer as adjunct to a subculturing exercise. The following review information may assist you in proceeding through the exercise.
1. Cell populations are usually expressed as # cells/ml or # cells/culture.
2. When viewed with a compound microscope with a 10x ocular and 10x objective (total magnification 100x), one large square of the hemocytometer (1 mm x 1 mm) will fill the field.
3. Each of the four large corner squares and the large center square of a Neubauer type hemocytometer usually are counted. When the cell count is low (less than 10 cells/square) all nine squares should be counted.
4. Each large square measures 1 x 1 mm and is 0.1 mm deep. Hence, each large square has a volume of 0.1 mm3 or 0.001 cm3 or 0.0001 ml (10-4 ml).
5. The final calculation takes into account:
a. how you wish to express your count (cells/ml or cells/flask) b. the dilution (# of mls of saline or dye or medium into which your cells have been suspended c. the number of squares counted
Hence the number of cells/ml of sample is calculated as follows:
total # cells counted x dilution x 104 number of squares counted
PROCEDURE:
1. Proceed to make a 1:3 split of a culture following the procedure described above. In order to determine the number of cells which were harvested from the flask.
2. Aseptically remove 0.5 ml of cell suspension in trypsin and place in a microfuge tube on ice. Sterility need not be maintained.
3. Remove a few drops of the cell suspension with a Pasteur pipette and load both chambers after putting the hemocytometer coverslip in place.
4. Count the total number of cells in each of the four corner and central squares.
CALCULATIONS:
1. Count 10 squares and then multiply that number by 1,000 and that gives us the number of cells per ml from your solution of cells in trypsin.
N = Sum of 10 Squares X 103
2. This gives you the number of cells per ml, you should now adjust this for any dilution you are doing. That is, you have a T-25 flask that you have just added 3.3 ml of trysin to, this is our total volume of cells and you have calculated the number of cells per ml. You have removed 0.3 ml (or whatever volume you have used), so you have 3.0 ml of cells left that you are going to add to a T-25 with 9.0 ml of fresh media. This gives you a 1:10 dilution of the cells you have just counted.
OK, we have some data, let's see how good it is. Or more accurately, how precise is our data. Precise is defined in a statistics as the closeness of repeated measurements. We will now load the hemocytometer and count 5 squares per side (total of 10 squares) two times.
This will give us two groups of 10 readings (each reading represents one square). I want to know if the means of these two groups of 10 readings are significantly different at the 0.05 level of significance (alpha is equal to 0.05). Generate a null hypothesis to test this notion and test for significance.
Slide I Slide II
1. 1.
2. 2.
3. 3.
4. 4.
5. 5.
6. 6.
7. 7.
8. 8.
9. 9.
10. 10.
III. MATERIALS
Sterile ATTC growth media, PBS buffer and 0.25X trypsin in 100 ml media bottles (1 per group) Sterile 10 ml and 1 ml pipettes with battery – operated pippetors Pasture pipettes and bulbs T-25 tissue culture flasks Inverted phase contrast microscopes 28C CO2 Incubator Sterile laminar flow hood Hemocytometers Eppendorf pipettors and tips Coulter counter flasks and isoton solution Squirt bottles with 70% ethanol for sterile wipes, kimwipes 1000 ul and 100 ul eppendorffs with sterile tips
IV. REFERENCES
1. Jakoby, W. B. and Pastan, I. H. 1979. Cell Culture. Methods in Enzymology Volume LVIII. Academic Press, New York.
2. Freshney, R. I. 1987. Culture of Animal Cells: A Manual of Basic Technique. Alan R. Liss, Inc. Publishers, New York.
3. Gasque, C. E. 1989. A Manual of Laboratory Experiences in Cell Biology. W. C. Brown Publishers, Dubuque, Iowa.
4. Conn, P. M. 1990. Cell Culture. Academic Press, New York.
5. Much of what I have presented is taken from a course I had in In Vitro Toxicology at the Center For Advanced Training in Cell and Molecular Biology at Catholic University, Washington, D.C. 1986.
LAB 6 AND 7
IN VITRO INVESTIGATIONS OF ZF-4 CELLS
OK, we have hopefully mastered some skills in cell culture techniques and we have observed the growth of ZF-4 cells in culture. This weeks lab will be an experiment that your group has designed to study the effect of exogenous agents on ZF-4 cells. The students in each group must work together to design and perform your experiment. Remember, the design of your experiment MUST BE COMPLETED AND CLEARED WITH ME PRIOR THE SCHEDULED LAB (see syllabus). You will be provided with the following materials for the laboratory. Other materials not listed may be made available upon special request.
various drugs T-25 flasks MEM media with 10% sera Hanks buffer hemocytometers pipettes horse sera ethyl alcohol other materials are available upon request Sterile 1M HCl and 1M NaOH Ethanol, Caffeine
|